Learn more about Pharmacology
Pharmacology in 60 seconds
In this module, you will learn more about
- Pharmacokinetics: drug absorption, distribution, metabolism, excretion
- Pharmacodynamics: how drugs act within the body
Learn even more: See Chemistry and Cells
1 Pharmacokinetics: drug absorption, distribution, metabolism, excretion
Pharmacokinetics is the study of how a drug moves through the body system – what the body does to the drug.
1 Membrane transport
|
|
|
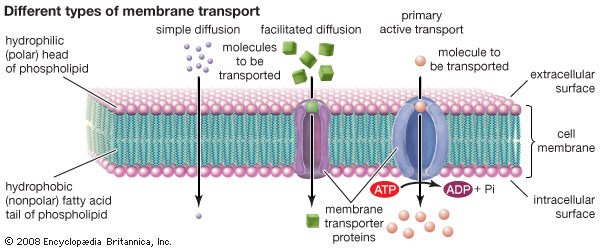
The cell membrane is a semipermeable structure that results in some molecules being able to passively diffuse across it whilst others are kept out and must be transported across it. The membrane is a phospholipid bilayer that allows lipid soluble molecules such as carbon dioxide CO2, to cross, whereas others such as sodium Na+ and potassium K+ can only leak across very, very slowly and so must be actively transported in and out of the cell. There are basically four ways in which molecules cross the membrane:
- Passive diffusion
- Facilitated diffusion
- Active transport
- Bulk transport
|
Passive diffusion
Where the molecules move from the side of the membrane with the highest concentration to the side which has the lowest concentration. That is, the molecules move down the concentration gradient created by the membrane barrier. Lipids can diffuse across the cell phospholipid membrane including the membranes of the blood brain barrier to enter the central nervous system. Polar molecules (ie those that dissolve in water such as electrolytes) cannot easily cross the phospholipid membrane. In passive diffusion solutes move down their concentration gradient to equalise the concentration on either side of a permeable membrane. |
|
|
Facilitated diffusion
Where a transport protein embedded in the wall of the membrane allows passage through the membrane. Polar molecules need to be transported from the outside to the inside of the cell. A number of mechanisms exist to allow this process:
|
|
|
Active transport
Where energy (ATP) is required to move molecules across the membrane against the concentration gradient. solutes move against the concentration gradient. In primary active transport ATP is utilised to pump ions against the gradient; in secondary active transport and co-transporter is used to move ions against the concentration gradient. |
|
Bulk transport
endocytosis (movement into the cell) and exocytosis (movement out of the cell).
endocytosis (movement into the cell) and exocytosis (movement out of the cell).
2 Ionisation of molecules
Ionisation is the addition or removal of an electron to create an ion.
Salt molecules are held together with ionic bonds where electrons are exchange between the element atoms. When we place a salt into water, the polar water molecules cause the salt molecule to separate into ions. For example, table salt which is sodium chloride, when placed into water, becomes sodium ions and chloride ions. Ions carry a charge that reflects the number of electrons that are in the outer electron orbit (see chemistry topic). Losing an electron creates a positive ion; gaining an electron creates a negative ion.
NaCl + H2O -> Na+ + Cl-
Since ions carry a charge they are repelled by other molecules that carry a similar charge. Recall the phospholipid bilayer that makes up the cell membrane. This membrane repels these charged ions, keeping sodium ions either inside or outside of the membrane. This means that sodium ions have to be pumped across the membrane by the sodium –potassium-ATPase pump.
Similarly if and acid is placed into an alkaline solution the acid becomes ionised whereas an acid in an acid solution does not. If an alkaline is placed into acid solution, the alkaline one will become ionised. Once a molecule is ionised it is more difficult for the ion to cross the cell membrane.
Salt molecules are held together with ionic bonds where electrons are exchange between the element atoms. When we place a salt into water, the polar water molecules cause the salt molecule to separate into ions. For example, table salt which is sodium chloride, when placed into water, becomes sodium ions and chloride ions. Ions carry a charge that reflects the number of electrons that are in the outer electron orbit (see chemistry topic). Losing an electron creates a positive ion; gaining an electron creates a negative ion.
NaCl + H2O -> Na+ + Cl-
Since ions carry a charge they are repelled by other molecules that carry a similar charge. Recall the phospholipid bilayer that makes up the cell membrane. This membrane repels these charged ions, keeping sodium ions either inside or outside of the membrane. This means that sodium ions have to be pumped across the membrane by the sodium –potassium-ATPase pump.
Similarly if and acid is placed into an alkaline solution the acid becomes ionised whereas an acid in an acid solution does not. If an alkaline is placed into acid solution, the alkaline one will become ionised. Once a molecule is ionised it is more difficult for the ion to cross the cell membrane.
3 Promoting chemical reactions and the role of enzymes
For chemical reactions to take place in the body the molecules that need to interact need to be, firstly, in the right place and, secondly, in the right concentrations.
|
Many reactions in the body are simply too slow for healthy active living and so these reactions are facilitated by biological enzymes. In chemistry these are named catalysts. The enzyme speeds up the reaction rate by lowering the amount of energy required for the reaction.
Enzymes are specific for each chemical process whether it is to break down a molecule into metabolites or to build a new molecule such as a protein. Structurally enzymes are proteins and so are susceptible to being destroyed by acids or high body temperature. Enzymes are not used up during the chemical reaction. They often require cofactors (eg inorganic minerals) and coenzymes (eg organic vitamins) to function correctly. |
|
|
Enzymes can be important drug targets.
An example is angiotensin converting enzyme (ACE). ACE causes angiotensin 1 to be converted to angiotensin 2. Angiotensin 2 is a powerful vasoconstrictor and so has a role in controlling blood pressure. It also causes the adrenal gland to release aldosterone which increases renal sodium retention which causes increased water retention leading to an increase in blood volume, adding to a rise in blood pressure. The trigger for the enzyme to cause this conversion of angiotensin 1 to angiotensin 2 is a drop in blood pressure detected by the kidneys which then release renin. Drugs known as angiotensin converting enzyme inhibitors (ACEIs) prevent the enzyme from enabling the production of angiotensin 2 resulting in the blood pressure returning to normal in those with high blood pressure (see flowchart). |
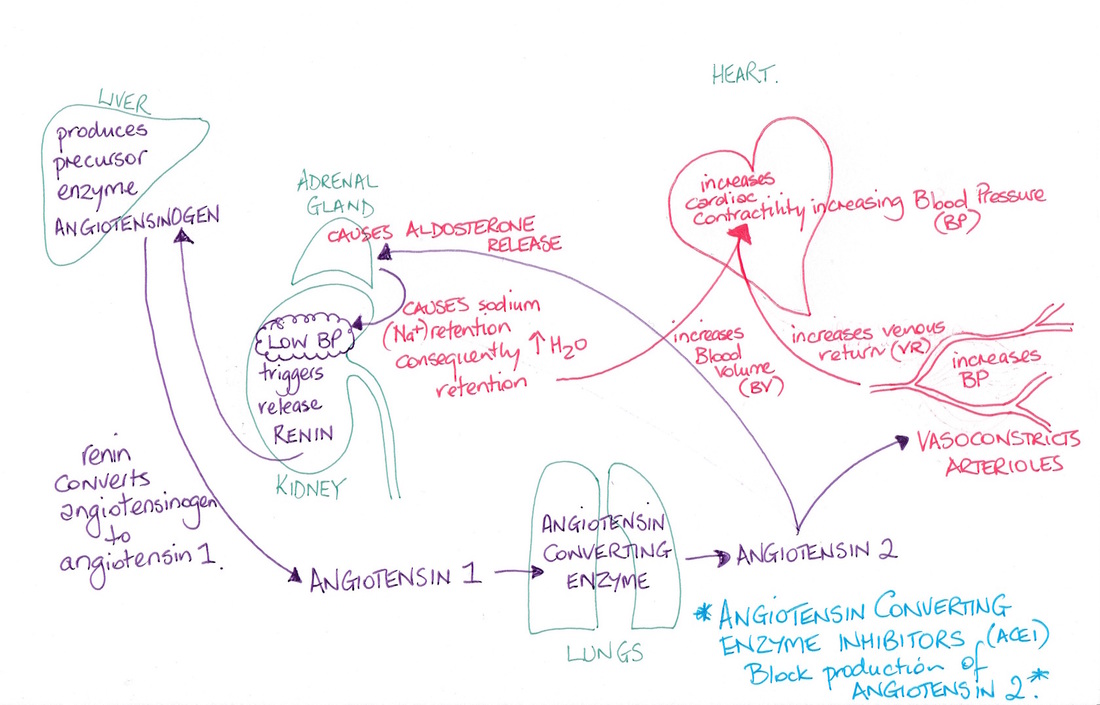
© Dr Patricia Logan 2016 - Renin Angiotensin–Aldosterone System (RAAS) flowchart
|
4 Absorption, distribution, metabolism and excretion of a drug
|
Absorption and distribution
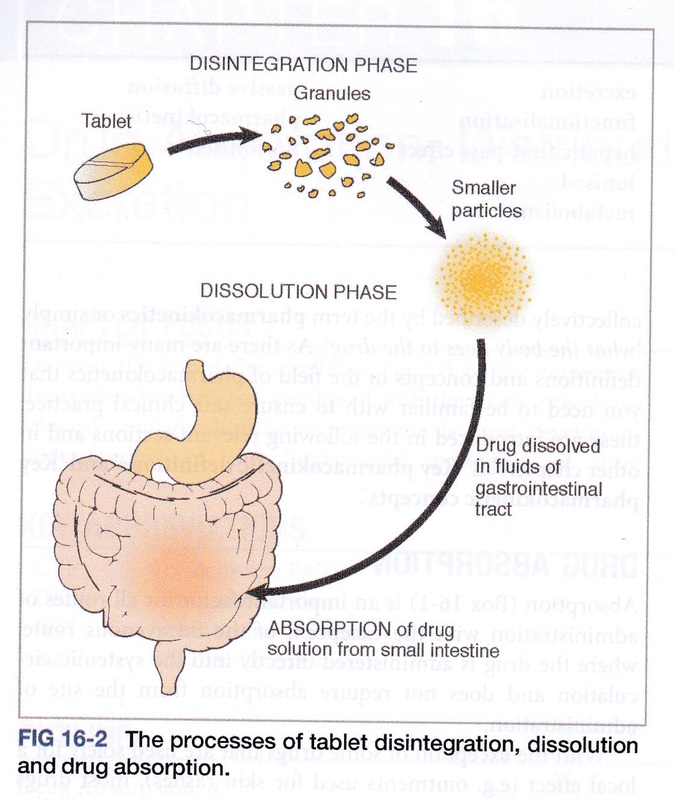
When a drug is given orally it has to be absorbed from the gastrointestinal tract (GIT) and enter then blood stream. Intravenous drugs bypass the GIT entirely and intramuscular injected drugs take advantage of the high blood flow of the muscles to deliver the drug to the systemic blood supply. The dense capillary networks in the rectal walls or those under the tongue enable drug absorption, if lipid soluble, directly to the systemic blood and so avoiding the GIT. The stomach is highly acidic which can destroy some drugs especially if they are proteins in structure. In theory, drugs that are acids will cross the cell membranes more readily from the stomach, since an acid in an acid does not ionise. An example would be aspirin which is an acid. However, the stomach does not have a large surface area for absorption of drugs. The small bowel however, does have a very large surface area and the environment is alkaline thanks to the delivery of bicarbonate ions from the pancreas into the duodenum. For this reason some drugs are covered in a coating (termed enteric coating) to prevent stomach acid damaging the chemical structure before the drug reaches the duodenum. Going back to our example of aspirin, since it is an acid, it will ionise in the alkaline environment of the duodenum hindering its absorption across the membrane – however, enough of the drug will cross to have a therapeutic effect due to the very large surface area. Some drugs used to treat GIT microbial infections are useful because they are not absorbed and consequently remain in the GIT lumen. These are excreted in the faeces. Blood from the GIT travels first to the liver via the porta hepatitis and then leaves the liver via the hepatic vein returning to the heart from where it is then pumped around the body to it reach its target. Where the drug travels to is described as the drug distribution. The drug will not only travel to the target. Where it is distributed to will depend on its chemical composition and it ability to cross cell membranes as well as the blood tissue perfusion. A proportion of drug molecules, upon reaching the blood plasma, will bind to proteins and lipoproteins, usually reversibly. Acidic drugs upon entering the systemic circulation tend to bind to albumin. An example of highly protein bound drug is warfarin. Only the molecules of the drug that are not bound to albumin can have a therapeutic effect. Warfarin, an anticoagulant, is 99% bound with only 1% of the drug not bound, that is, free to be distributed to tissues. As the binding process is reversibly, as drug leaves the blood to enter the tissues, more drug is released from the albumin to become free for distribution. Drugs can compete for binding sites, influencing the bound and free portions of the drug particularly if the excretion rate is compromised. There are barriers to drug distribution from the systemic circulation such as the blood brain barrier. If a drug is lipid soluble it can cross the blood brain barrier and other physiological barriers such as the placental barrier, but a polar drug is much less likely to cross the membranes. Examples of this can be seen with the anti-histamines or nasal decongestants included in cold and flu tablets. The daytime tablet includes a nasal decongestant that cannot cross the blood brain barrier and so has less sleep effect whereas the night-time tablet may contain one that can cross to help with sleep. However, some drugs are contained by the compartments they are injected into for example, if a drug is injected into a synovial joint, the synovial capsule may contain it restricting the drug to the joint capsule. The ideal situation would be that a drug only concentrated at the site of the drug target. However, in reality, that does not happen. Drugs can concentrate in varying amounts in the body’s diverse tissues. When a drug is being trialled, the amount of drug that concentrates in the major organs and the brain is measured. These measurements indicate the maximum concentration of the drug in an organ before damage is caused in that organ. The organ may not be the target organ, and so this value will limit the size of the dose of the drug that can be given. |
|
|
First pass metabolism
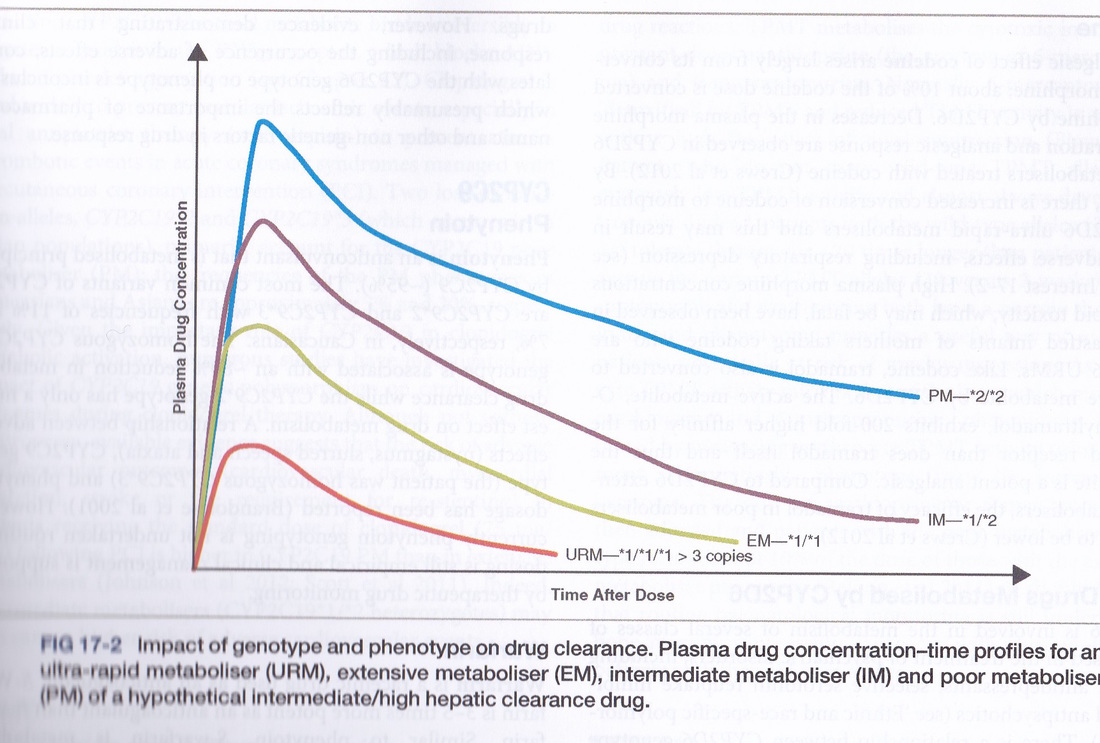
While the drug is in the liver, hepatic cell processes may act on the drug. This is named hepatic or liver metabolism. Drug metabolism can occur elsewhere in other tissues however the main metabolism of drugs occurs in the liver by the actions of a group of enzymes known as the P450 enzymes. Liver metabolism renders non polar (lipid soluble) drugs more water soluble so that the kidneys can more readily excrete them. Consequently a drug that is given orally may not reach the systemic blood circulation intact, with only a percentage of the original drug dose becoming available to the tissues. Metabolism may result in active drug products (metabolites) that may be less or more biologically active than the original drug. Other drugs do not undergo any liver metabolism. The size of oral drug doses are calculated to compensate for the hepatic metabolism. Since enzymes are proteins, the cytochrome P450 enzymes are produced according to an individual’s inherited DNA codes (P450 enzymes are symbolised as CYP and then followed by a distinguishing code eg CYP1A2). This means that not every person will have the exact same set of enzymes – there are variants, many of which are now known and many which are still being discovered. An example of genetic variation in P450 enzymes is the enzyme that acts on codeine. Each person inherits one allele for a protein from their father and one from their mother resulting in either a homologous (two the same) or heterozygous (two different) alleles in the child’s DNA codes. This is known as the genotype (See the Inheritance topic). How that allele pair is expressed in a person is called the phenotype. In the case of how a drug affects a person the phenotype is the clinical manifestation. Some patients will complain that the drug codeine does not help their pain level and if this is the case it is likely they have the allele pair that results in them being a poor metaboliser for the drug. Codeine is converted to morphine by liver enzymes. Failure to convert the drug to morphine will result in a suboptimal therapeutic effect. Allele variants result in people being categorised as poor metabolisers (PM), intermediate metabolisers (IM) , extensive metabolisers (EM) or ultra-rapid metabolisers (URM) for a number of drugs. Someone who is a poor metaboliser will excrete a lipid soluble drug more slowly than a person who is an extensive metaboliser for that drug. Mechanisms for avoiding the first pass effect include giving the drug in ways other than oral administration, for example, intravenously, intramuscularly, absorption from under the tongue or from the rectum. In all these methods the drug enters the systemic circulation directly. In the case of intravenous injections the bioavailability of the drug is 100%. In these cases the drug circulates through the body avoiding the liver and so reaches the drug target before being metabolised. |

|
Drug excretion
Drugs can be excreted in sweat, saliva, mucous, bile and faeces, and even breathed out such as the anaesthetic halothane. Ethanol, which is a volatile chemical (moves from liquid to gas easily) is highly soluble in blood and is excreted in limited amounts by the lungs. This forms the basis of the alcohol breathe test. However, the main organs of excretion are the kidneys. Some drugs are excreted unchanged in urine whilst others are almost completely metabolised so that very little of the original drug is detected in urine samples. Glomerular filtration and tubular secretion facilitate the movement of drugs and their metabolites into the filtrate that becomes the urine. All drugs undergo some level of filtration as the systemic circulation passes through the kidneys. Lipophilic drugs can be reabsorbed by tubular resorption back to the systemic circulation.
Drugs excreted by the kidneys need to be water soluble so that they are not reabsorbed from the filtrate. Liver metabolism converts lipid soluble drugs to more water soluble chemicals aiding in their excretion by the kidneys. Kidneys begin to lose nephrons after the age of 35 years and so renal function begins a very slow decline. Functioning nephrons enlarge to compensate for those that are lost and so renal failure may not be detected until very late in the process of decline. Some drugs damage nephrons inducing renal failure; others create damage only if the therapeutic concentrations are exceeded. An example of a drug where renal output should be monitored is gentamicin, an intravenous antibiotic. This drug has a low therapeutic index (see therapeutic range below).
Urinary pH will also effect the amount of drug that is reabsorbed by altering the amount of ionisation. Weak acids are excreted more readily in weak alkaline solution but more slowly in acidic urine (see notes on ionisation above); the reverse is true for weak bases. By changing the pH of the urine excretion can be increased or decreased.
Drugs excreted in bile once in the gut lumen, may be reabsorbed into the blood circulation. This is referred to as enterohepatic recycling. The gut normal flora bacteria in some cases can reform the active drug. For drugs where this is the case the drug dosages take this recycling into account. An example is the oral contraceptive pill (OCP). It means that any disruption to the bowel flora (such as diarrhoea or the use of broad spectrum anti-biotics) can alter the concentration of the drug in the systemic circulation decreasing the drug concentration and limiting the effect of the drug. In the case of the OCP it may result in an unexpected pregnancy.
Drugs can be excreted in sweat, saliva, mucous, bile and faeces, and even breathed out such as the anaesthetic halothane. Ethanol, which is a volatile chemical (moves from liquid to gas easily) is highly soluble in blood and is excreted in limited amounts by the lungs. This forms the basis of the alcohol breathe test. However, the main organs of excretion are the kidneys. Some drugs are excreted unchanged in urine whilst others are almost completely metabolised so that very little of the original drug is detected in urine samples. Glomerular filtration and tubular secretion facilitate the movement of drugs and their metabolites into the filtrate that becomes the urine. All drugs undergo some level of filtration as the systemic circulation passes through the kidneys. Lipophilic drugs can be reabsorbed by tubular resorption back to the systemic circulation.
Drugs excreted by the kidneys need to be water soluble so that they are not reabsorbed from the filtrate. Liver metabolism converts lipid soluble drugs to more water soluble chemicals aiding in their excretion by the kidneys. Kidneys begin to lose nephrons after the age of 35 years and so renal function begins a very slow decline. Functioning nephrons enlarge to compensate for those that are lost and so renal failure may not be detected until very late in the process of decline. Some drugs damage nephrons inducing renal failure; others create damage only if the therapeutic concentrations are exceeded. An example of a drug where renal output should be monitored is gentamicin, an intravenous antibiotic. This drug has a low therapeutic index (see therapeutic range below).
Urinary pH will also effect the amount of drug that is reabsorbed by altering the amount of ionisation. Weak acids are excreted more readily in weak alkaline solution but more slowly in acidic urine (see notes on ionisation above); the reverse is true for weak bases. By changing the pH of the urine excretion can be increased or decreased.
Drugs excreted in bile once in the gut lumen, may be reabsorbed into the blood circulation. This is referred to as enterohepatic recycling. The gut normal flora bacteria in some cases can reform the active drug. For drugs where this is the case the drug dosages take this recycling into account. An example is the oral contraceptive pill (OCP). It means that any disruption to the bowel flora (such as diarrhoea or the use of broad spectrum anti-biotics) can alter the concentration of the drug in the systemic circulation decreasing the drug concentration and limiting the effect of the drug. In the case of the OCP it may result in an unexpected pregnancy.
5 Bioavailability
Bioavailability is the amount of the drug that reaches the blood plasma. If a drug is given intravenously then the bioavailability is 100%. However, an oral dose of a drug may undergo the first pass effect resulting in a bioavailability of less than 100%. Liver metabolism can render a drug useless in which case the drug is only given by parenteral routes (intravenous, sublingual, rectal, intramuscular). Liver disease can alter the capacity of the liver to produce plasma proteins and so this can have effect on the bound versus free portions of a protein bound drug.
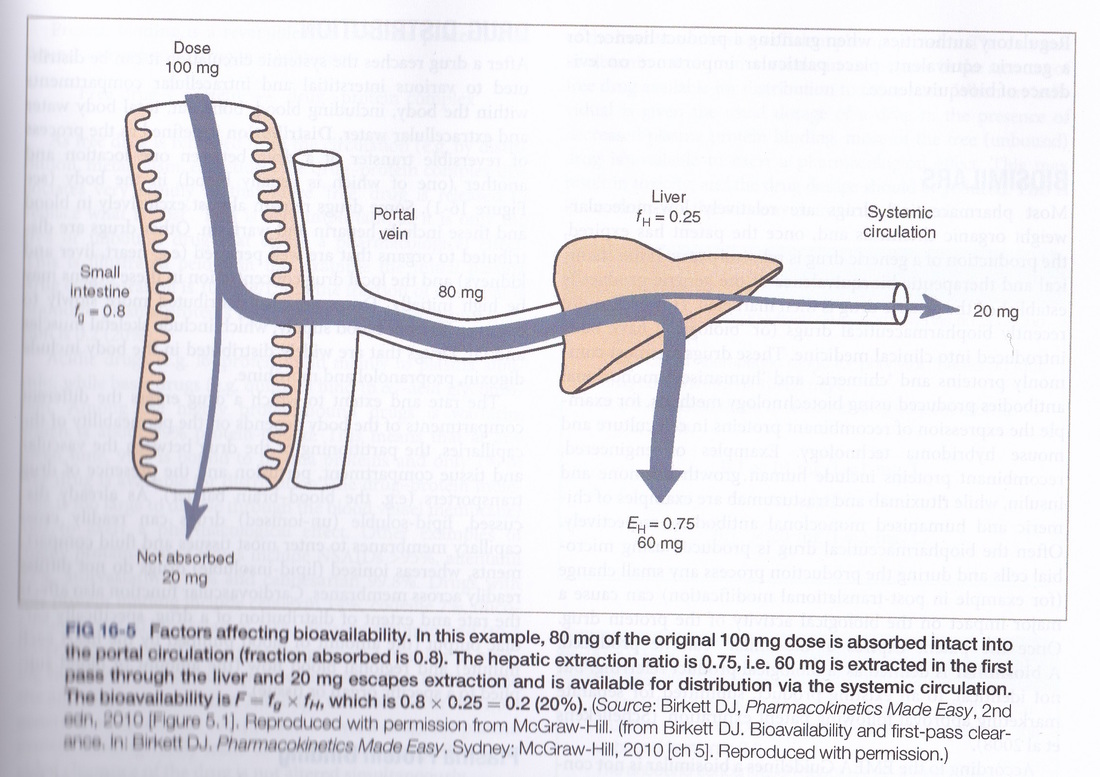
6 Biological half-life
Biological half-life indicates how long a chemical will remain in the body with respect to normal absorption and excretion rates. We can measure the amount of drug in the plasma over time by taking blood samples. This tells us how long it takes for a drug to reach the plasma through absorption and how long it takes to be removed from the plasma. The drug half-life is important for determining dose timing in order to maintain a therapeutic concentration of the drug whilst minimising concentration troughs and peaks.
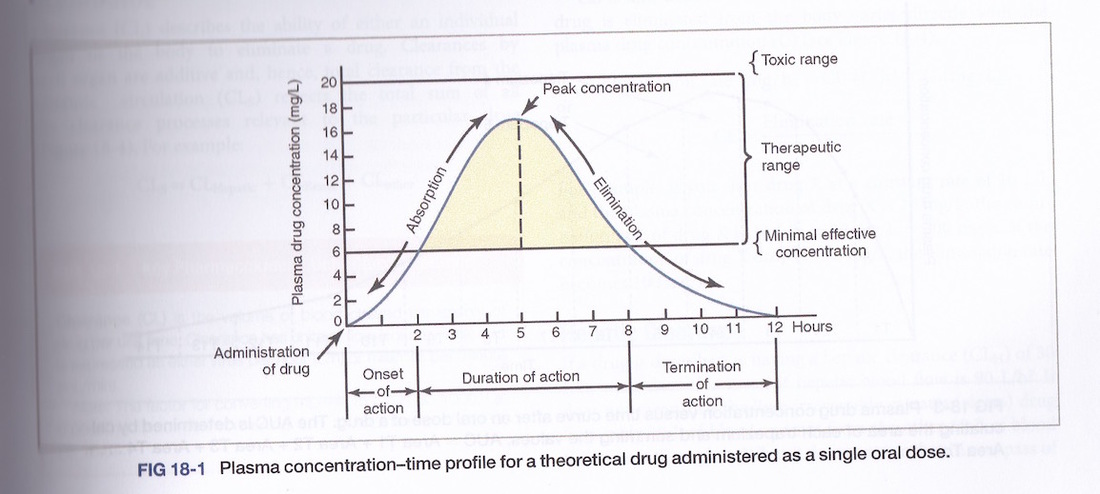
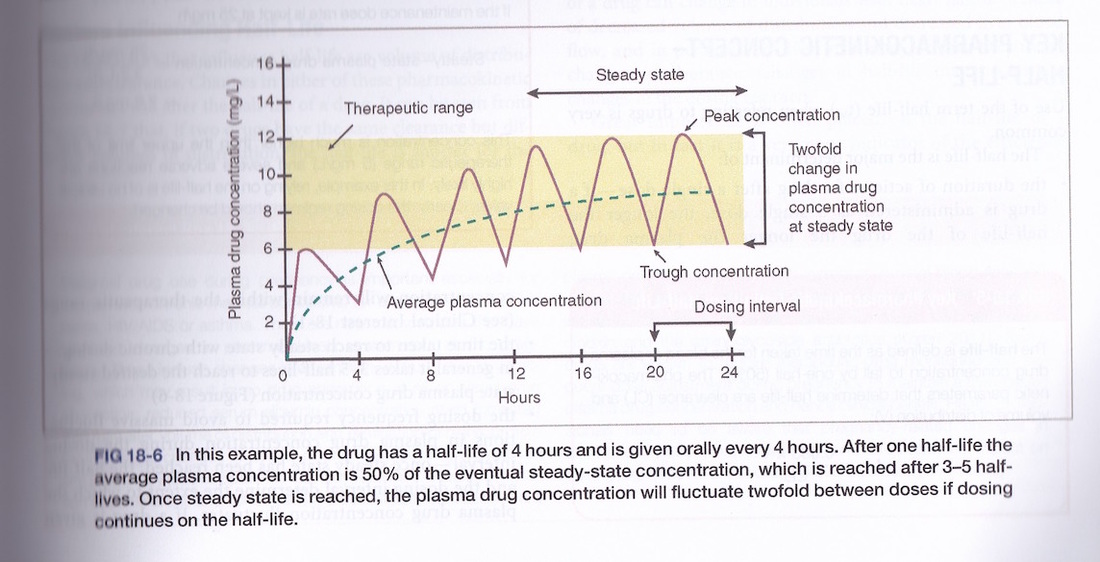
7 Therapeutic range
The concentration of the drug can be measured using blood samples whereas it is difficult to measure it in tissues. For a very few drugs it is possible to individualise the dosing regime in order to achieve an effective drug plasma concentration. The therapeutic range for a specific drug indicates the range of plasma concentrations that have a high probability of producing a therapeutic effect whilst having a low probability of creating adverse events. The therapeutic range might be very narrow such as for Lithium (used in the treatment of manic depression) or very broad such as with morphine.
Therapeutic index (TI): indicates the limitations of the drug dosage with respect to relative effective dose to achieve a therapeutic effect compared to the dose that induces toxicity. Essentially a measure of efficacy versus safety. Lithium has a TI of 4:1 indicating it has to be regular monitored to make certain the plasma concentration levels do not exceed the therapeutic range. Other examples of drugs with narrow a therapeutic index include warfarin, paracetamol (acetaminophen), theophylline. In comparison, morphine has an index of 70:1 (Stanley, T. (2000). Anaesthesia for the 21st century. Proc. Bayl. Univ. Med. Cent. 13(1): 7-10. PMID 16389318).
Therapeutic index (TI): indicates the limitations of the drug dosage with respect to relative effective dose to achieve a therapeutic effect compared to the dose that induces toxicity. Essentially a measure of efficacy versus safety. Lithium has a TI of 4:1 indicating it has to be regular monitored to make certain the plasma concentration levels do not exceed the therapeutic range. Other examples of drugs with narrow a therapeutic index include warfarin, paracetamol (acetaminophen), theophylline. In comparison, morphine has an index of 70:1 (Stanley, T. (2000). Anaesthesia for the 21st century. Proc. Bayl. Univ. Med. Cent. 13(1): 7-10. PMID 16389318).
8 How drugs can interact with cells
|
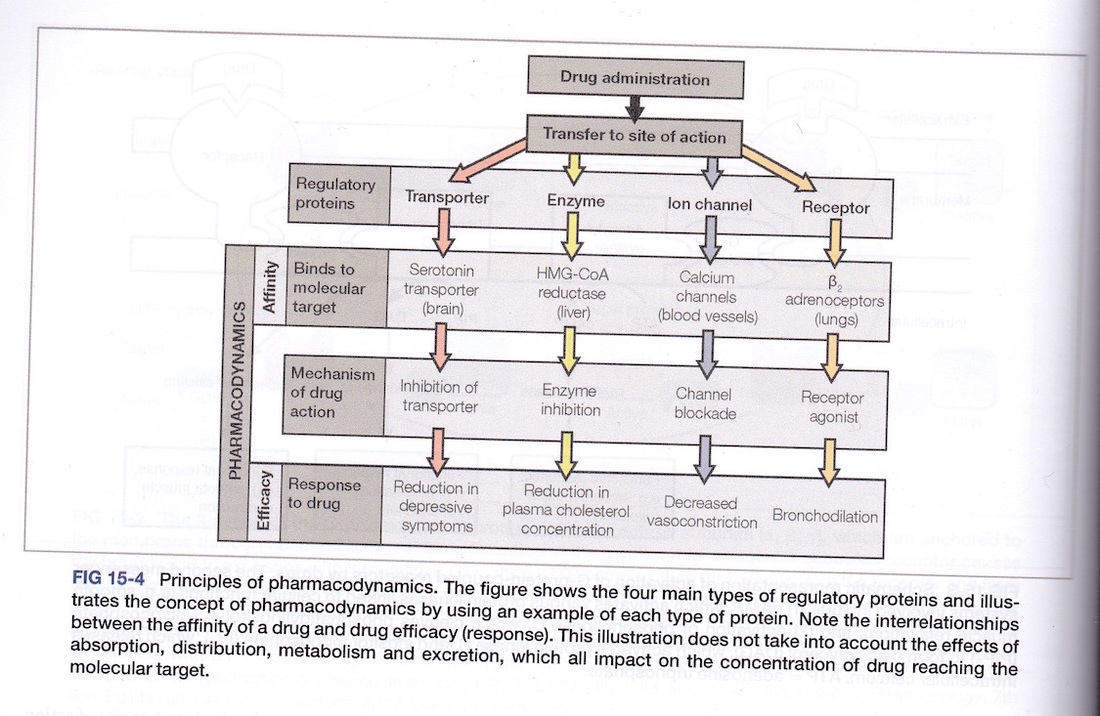
The study of how drugs interact with cells and their targets is referred to as pharmacodynamics. A few examples have been outlined in the sections above.
|
|
|
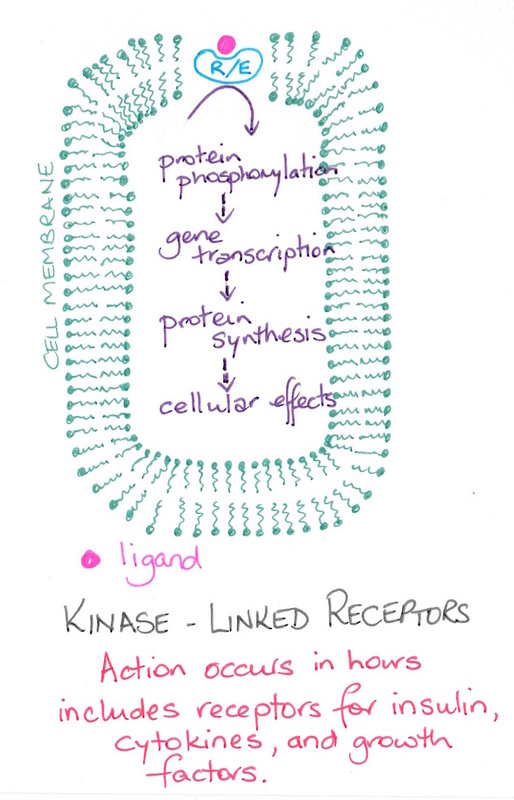
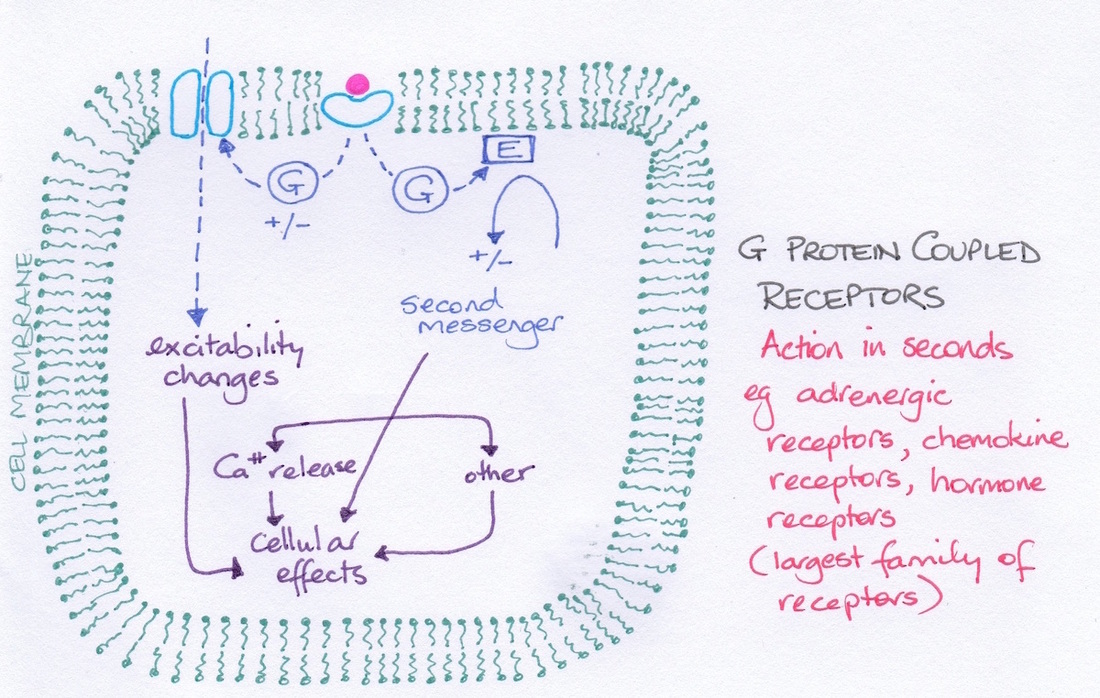
In order to understand how drugs reach their targets we first need to look at the types of targets available for drug interaction. Basically there are four groups of targets: receptors, ion channels, enzymes and transporter proteins (see diagram). With respect to receptors we need to examine effector- receptor linkages (see diagram).
|
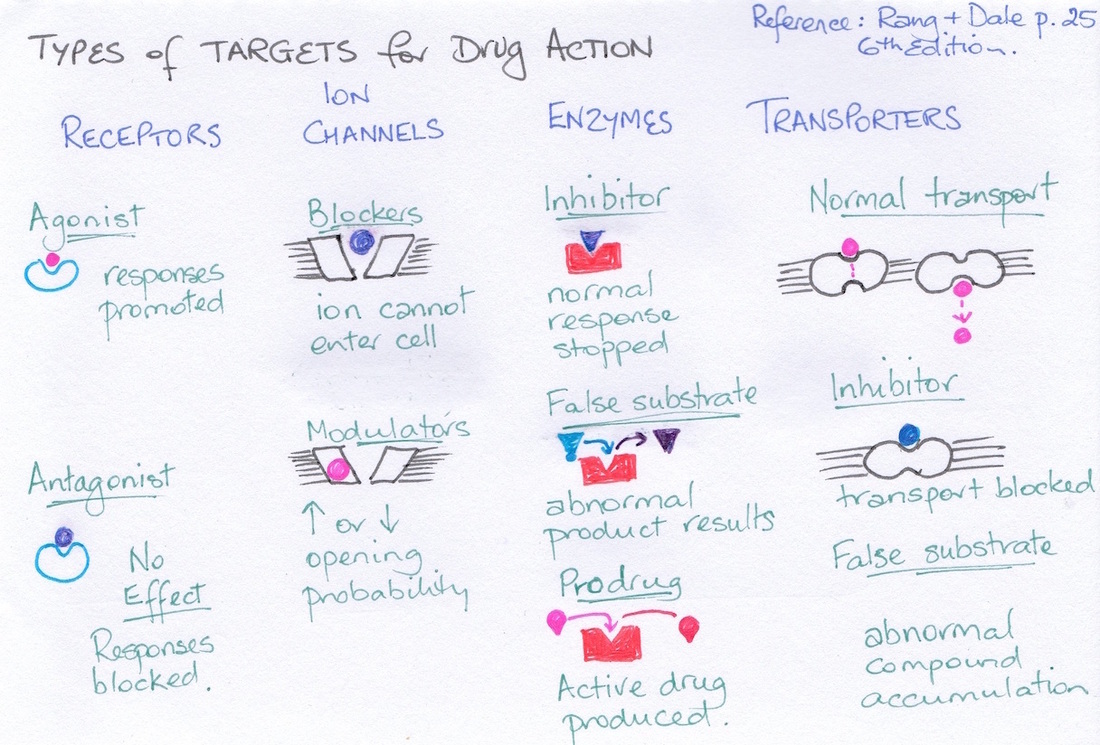
|

9 Agonist and antagonist drugs
Chemicals that interact with these targets may bind them preventing the endogenous chemical from binding to create an effect; these are termed antagonists and sometimes simply described a blockers such as calcium channel blockers (block voltage gated calcium channels) or beta blockers (block adrenergic receptors). Most drugs, if antagonists, are reversible antagonists. And so the effect of the drug diminishes as the concentration of the drug declines with excretion. However, an irreversible antagonist stays in place until such time as the protein it is attached to is removed and recycled.
Those drugs that initiate an effect when attaching to a receptor are referred to as agonists.
Drug that compete for binding sites can be pushed off the binding by a drug with a higher affinity for the site. The example often given is the opioid drug morphine which binds to opioid receptors inhibiting pain transmission. The drug naloxone is used to treat opioid drug overdose because it competes with the morphine for the opioid binding sites, pushing it off the receptors and so reversing the effect of the morphine. Morphine is respiratory depressant (among its many other effects) and so in overdose can result in respiratory failure.
The figure below outlines the mechanisms for receptor agonists and antagonists
Those drugs that initiate an effect when attaching to a receptor are referred to as agonists.
Drug that compete for binding sites can be pushed off the binding by a drug with a higher affinity for the site. The example often given is the opioid drug morphine which binds to opioid receptors inhibiting pain transmission. The drug naloxone is used to treat opioid drug overdose because it competes with the morphine for the opioid binding sites, pushing it off the receptors and so reversing the effect of the morphine. Morphine is respiratory depressant (among its many other effects) and so in overdose can result in respiratory failure.
The figure below outlines the mechanisms for receptor agonists and antagonists

Bibliography
Bryant & Knights (2015). Pharmacology for Health Professionals (4th Edn). Elsievier.
Rang, Dale, Ritter & Flower (2007). Rang and Dale’s Pharmacology (6th Edn). Churchill Livingstone.
Rang, Dale, Ritter & Flower (2007). Rang and Dale’s Pharmacology (6th Edn). Churchill Livingstone.

